Qu’est-ce que la fibrose kystique?
La fibrose kystique est une maladie héréditaire due à une mutation (ou erreur) du gène du régulateur de conductance transmembranaire de la fibrose kystique (CFTR).

| La fibrose kystique est une maladie génétique héréditaire, qui se transmet selon un mode autosomique récessif. Cela signifie qu’un bébé ne naîtra avec la fibrose kystique que s’il hérite de deux gènes CFTR défectueux – un de chaque parent. La fibrose kystique n’est pas contagieuse, et vous ne pouvez pas la développer. Si vos deux parents sont porteurs du gène CFTR défectueux, il y a 25 % (1 sur 4) des chances, que vous naissiez avec la fibrose kystique. Les hommes et les femmes sont tout aussi susceptibles d’être atteints de FK. Cette affection touche les personnes de toutes les origines ethniques, bien qu’elle soit plus fréquente dans la population de race blanche3. Une personne possédant un gène CFTR normal et un gène CFTR défectueux est dite porteuse de la fibrose kystique. Les porteurs ne sont pas atteints de fibrose kystique et ne présentent aucun des symptômes de la maladie. Les porteurs de la FK peuvent cependant, transmettre leur copie du gène CFTR défectueux à leurs enfants. Les tests génétiques permettent de savoir si vous êtes porteur du gène CFTR défectueux. |
Effets du gène CFTR
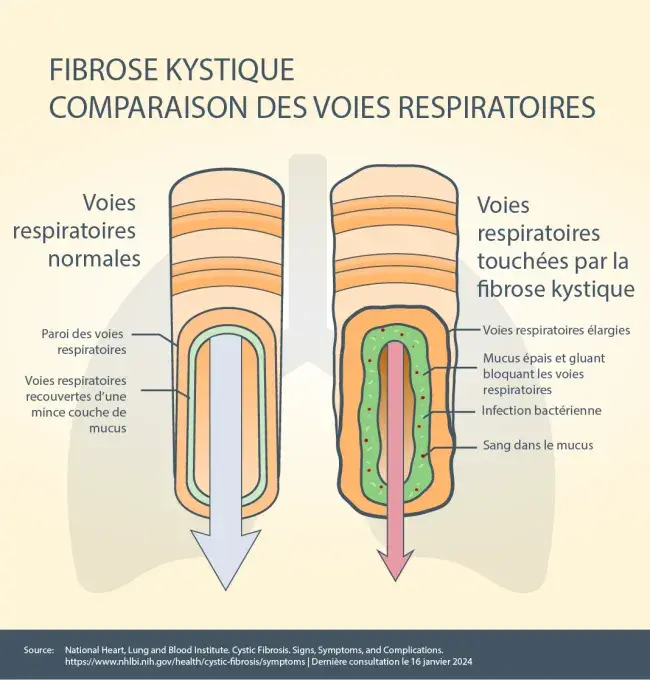
Le gène CFTR entraîne un dysfonctionnement de la protéine CFTR.La protéine CFTR se trouve dans tous les organes du corps, qui produisent du mucus, notamment les poumons, le pancréas, le foie et les intestins, ainsi que les glandes sudoripares. La protéine CFTR agit comme un canal de chlorure – elle contribue à maintenir l’équilibre entre le chlorure (un composant du sel) et l’eau à la surface de la cellule. Lorsque la protéine CFTR ne fonctionne pas correctement, elle est incapable d’aider à déplacer le chlorure vers la surface de la cellule. Sans chlorure pour attirer l’eau, votre mucus devient épais et collant et peut s’accumuler, ce qui entraîne des blocages, des lésions et des infections.
|
|