What causes Cystic Fibrosis?
Cystic fibrosis is a hereditary disease caused by a mutation (or error) in the cystic fibrosis transmembrane conductance regulator (CFTR) gene.
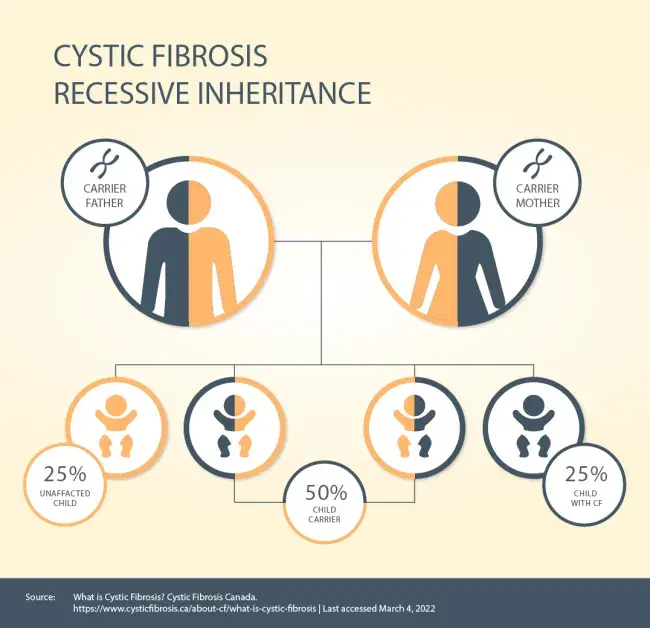
| Cystic fibrosis is inherited in an autosomal recessive pattern. This means a baby will only be born with cystic fibrosis if they inherit two defective CFTR genes – one from each parent. Cystic fibrosis isn’t contagious, and you can’t develop it. If both your parents carry the defective CFTR gene, there’s a 25% (1-in-4) chance that you’ll be born with cystic fibrosis. Males and females are equally as likely to have CF. The condition affects all ethnic backgrounds although it is more common in Caucasians.3 Someone with one normal CFTR gene and one faulty CFTR gene is known as a cystic fibrosis carrier. Carriers do not have cystic fibrosis and don’t exhibit any of the symptoms of the disease. CF carriers can pass their copy of the defective CFTR gene on to their children. Genetic testing can tell if you have the faulty CFTR gene. |
Effects of the CFTR Gene
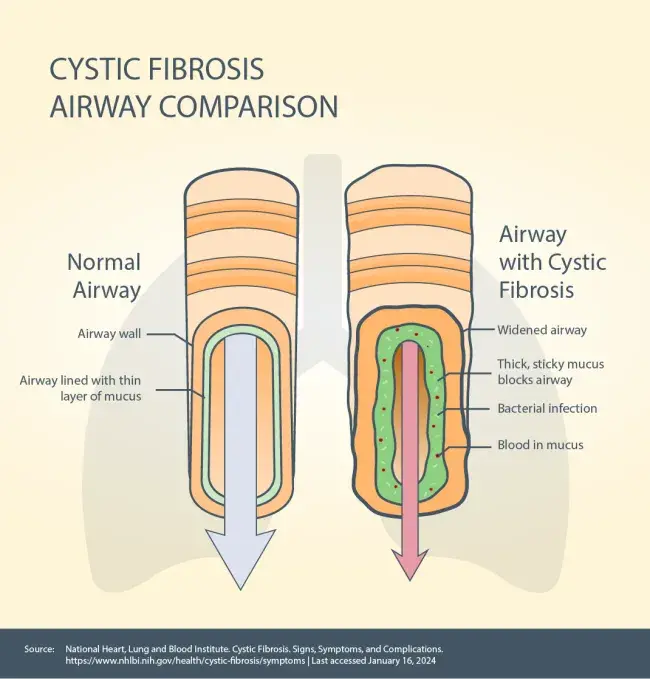
The CFTR gene causes the CFTR protein to become dysfunctional.The CFTR protein is in every organ of the body that makes mucus including your lungs, pancreas, liver and intestines, as well as your sweat glands. The CFTR protein acts as a chloride channel – it helps maintain the balance of chloride (a component of salt) and water on the cell’s surface. When the CFTR protein is not working properly, it’s unable to help move chloride to the cell’s surface. Without chloride to attract water, your mucus becomes thick and sticky and can build up – leading to blockages, damage and infections.
|
|